Fibrose quística

A fibrosis quística é unha enfermidade que provoca a acumulación de moco espeso e pegañento nos pulmóns, no tracto dixestivo e noutras áreas do corpo. É unha das enfermidades pulmonares crónicas máis comúns en nenos e adultos novos. É un trastorno potencialmente mortal.
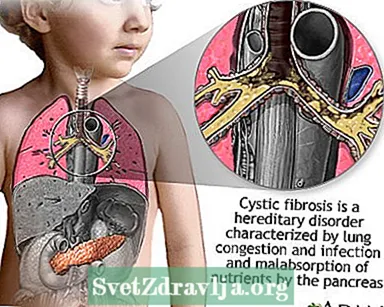
A fibrosis quística (FQ) é unha enfermidade que se transmite polas familias. É causada por un xene defectuoso que fai que o corpo produza fluído anormalmente espeso e pegañento, chamado moco. Este moco acumúlase nas vías respiratorias dos pulmóns e no páncreas.
A acumulación de moco provoca infeccións pulmonares que poñen en risco a vida e problemas graves de dixestión. A enfermidade tamén pode afectar ás glándulas sudoríparas e ao sistema reprodutivo do home.
Moita xente leva un xene CF pero non presenta síntomas. Isto débese a que unha persoa con FQ debe herdar 2 xenes defectuosos, un de cada pai. Algúns estadounidenses teñen o xene CF. É máis común entre os de orixe norte ou centroeuropa.
A maioría dos nenos con FQ son diagnosticados á idade de 2 anos, especialmente porque se realiza un exame de recén nacido en todos os Estados Unidos. Para un número pequeno, a enfermidade non se detecta ata os 18 anos ou máis. Estes nenos adoitan ter unha forma máis leve da enfermidade.
Os síntomas nos recentemente nados poden incluír:
- Crecemento atrasado
- Non engordar normalmente durante a infancia
- Non hai movemento intestinal nas primeiras 24 a 48 horas de vida
- Pel de sabor salgado
Os síntomas relacionados coa función intestinal poden incluír:
- Dor de barriga por estreñimiento severo
- Aumento do gas, inchazo ou barriga que aparece inchada (distendida)
- Náuseas e perda de apetito
- As feces pálidas ou de cor arxilosa, con mal olor, con moco ou que flotan
- Perda de peso
Os síntomas relacionados cos pulmóns e os seos poden incluír:
- Tose ou aumento do moco nos seos ou nos pulmóns
- Fatiga
- Conxestión nasal causada por pólipos nasais
- Episodios repetidos de pneumonía (os síntomas de pneumonía en alguén con fibrosis quística inclúen febre, aumento da tose e falta de aire, aumento do moco e perda de apetito)
- Dor ou presión nos seos causada por infección ou pólipos
Síntomas que se poden notar máis tarde na vida:
- Infertilidade (en homes)
- Inflamación repetida do páncreas (pancreatite)
- Síntomas respiratorios
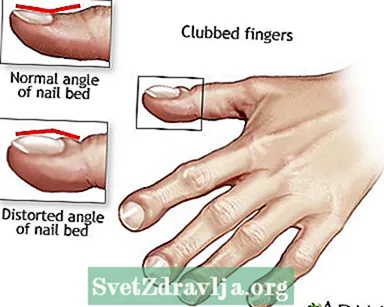
- Dedos achafarrados
Faise unha análise de sangue para axudar a detectar a FQ. A proba busca cambios no xene CF. Outras probas usadas para diagnosticar a FQ inclúen:
- A proba de tripsinóxeno inmunorreactivo (IRT) é unha proba estándar de detección de recién nacidos para a FQ. Un alto nivel de IRT suxire unha posible FQ e require máis probas.
- A proba de cloruro da suor é a proba de diagnóstico estándar para a FQ. Un alto nivel de sal na suor da persoa é un sinal da enfermidade.
Outras probas que identifican problemas que poden estar relacionados coa FQ inclúen:
- Radiografía de tórax ou tomografía computarizada
- Proba de graxa fecal
- Probas de función pulmonar
- Medición da función pancreática (feces elastase pancreática)
- Proba de estimulación da secretina
- Tripsina e quimotripsina nas feces
- Serie GI superior e intestino delgado
- Cultivos pulmonares (obtidos por esputo, broncoscopia ou hisopo da garganta)
Un diagnóstico precoz de FQ e un plan de tratamento poden mellorar tanto a supervivencia como a calidade de vida. O seguimento e o seguimento son moi importantes. Cando sexa posible, débese recibir atención nunha clínica especializada en fibrosis quística. Cando os nenos alcanzan a idade adulta, deben trasladarse a un centro especializado en fibrosis quística para adultos.
O tratamento para problemas pulmonares inclúe:
- Antibióticos para previr e tratar infeccións pulmonares e dos seos. Pódense tomar por vía oral, ou administrados nas veas ou mediante tratamentos respiratorios. As persoas con FQ poden tomar antibióticos só cando sexan necesarias ou todo o tempo. As doses adoitan ser máis altas do normal.
- Medicamentos inhalados para axudar a abrir as vías respiratorias.
- Outros medicamentos que se administran mediante un tratamento respiratorio para diluír o moco e facilitar a tose son a terapia enzimática ADNse e as solucións salinas altamente concentradas (solución salina hipertónica).
- Vacina contra a gripe e vacina contra o polisacárido contra o pneumococo (PPV) anualmente (pregúntelle ao seu médico).
- O transplante de pulmón é unha opción nalgúns casos.
- Pode ser necesaria unha terapia de osíxeno a medida que a enfermidade pulmonar empeora.
Os problemas pulmonares tamén se tratan con terapias para diluír o moco. Isto facilita a tose do moco dos pulmóns.
Estes métodos inclúen:
- Actividade ou exercicio que fai respirar profundamente
- Dispositivos que se usan durante o día para axudar a limpar as vías respiratorias de exceso de moco
- Percusión torácica manual (ou fisioterapia torácica), na que un membro da familia ou un terapeuta baten lixeiramente o peito da persoa, as costas e a área baixo os brazos.
O tratamento dos problemas nutricionais e intestinais pode incluír:
- Unha dieta especial rica en proteínas e calorías para nenos maiores e adultos
- Encimas pancreáticos que axudan a absorber as graxas e as proteínas, que se toman con cada comida
- Suplemento vitamínico, especialmente vitaminas A, D, E e K.
- O seu provedor pode aconsellar outros tratamentos se ten feces moi duras
Ivacaftor, lumacaftor, tezacaftor e elexacaftor son medicamentos que tratan certos tipos de FQ.
- Melloran a función dun dos xenes defectuosos que causa a FQ.
- Ata o 90% dos pacientes con FQ e elixibles para un ou máis destes medicamentos só ou en combinación.
- Como resultado, hai menos acumulación de moco espeso nos pulmóns. Tamén se melloran outros síntomas de FQ.
A atención e seguimento no fogar deben incluír:
- Evitar fume, po, sucidade, fumes, produtos químicos domésticos, fume da cheminea e mofo ou mildeu.
- Dar moitos líquidos, especialmente a bebés e nenos cando fai calor, cando hai diarrea ou feces soltas ou durante unha actividade física extra.
- Exercicio físico 2 ou 3 veces por semana. Nadar, trotar e andar en bicicleta son boas opcións.
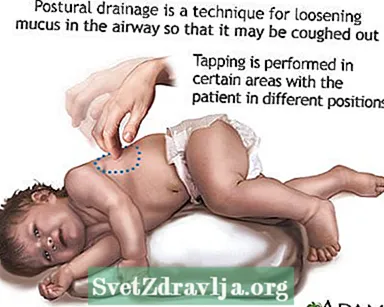
- Limpar ou traer moco ou secrecións das vías respiratorias. Isto debe facerse de 1 a 4 veces ao día. Os pacientes, as familias e os coidadores deben aprender a facer percusión no peito e drenaxe postural para axudar a manter as vías aéreas libres.
- Non se recomenda ningún contacto con outras persoas con FQ, xa que poden intercambiar infeccións (non se aplica aos membros da familia).
Pode aliviar o estrés da enfermidade uníndose a un grupo de apoio á fibrosis quística. Compartir con outras persoas que teñen experiencias e problemas comúns pode axudar a súa familia a non sentirse soa.
A maioría dos nenos con FQ mantéñense ben ata alcanzar a idade adulta. Son capaces de participar na maioría das actividades e asistir á escola. Moitos adultos novos con FQ rematan a universidade ou atopan emprego.
A enfermidade pulmonar empeora finalmente ata o punto en que a persoa está discapacitada. Hoxe en día, a vida media das persoas con FQ que viven ata a idade adulta é duns 44 anos.
A morte é máis frecuentemente causada por complicacións pulmonares.
A complicación máis común é a infección respiratoria crónica.
Outras complicacións inclúen:
- Problemas intestinais, como cálculos biliares, bloqueo intestinal e prolapso rectal
- Toser sangue
- Insuficiencia respiratoria crónica
- Diabetes
- Infertilidade
- Enfermidade hepática ou insuficiencia hepática, pancreatite, cirrose biliar
- Desnutrición
- Pólipos nasais e sinusite
- Osteoporose e artrite
- Pneumonía que non para de volver
- Pneumotórax
- Insuficiencia cardíaca cara dereita (cor pulmonale)
- Cancro colorrectal
Chame ao seu provedor se un bebé ou un neno ten síntomas de FQ e experimenta:
- Febre, aumento da tose, cambios no esputo ou sangue no esputo, perda de apetito ou outros signos de pneumonía
- Aumento da perda de peso
- Movementos intestinais ou feces máis frecuentes que cheiran mal ou teñen máis moco
- Barriga inchada ou aumento do inchazo
Chame ao seu provedor se unha persoa con FQ presenta novos síntomas ou se os síntomas empeoran, especialmente dificultades respiratorias graves ou tose de sangue.
Non se pode previr o CF. A detección de persoas con antecedentes familiares da enfermidade pode detectar o xene CF en moitos portadores.
CF
- Nutrición enteral - problemas de manexo do neno
- Tubo de alimentación de gastrostomía - bolo
- Como respirar cando che falta o alento
- Tubo de alimentación de xejunostomía
- Drenaxe postural
 Clubbing
Clubbing Drenaxe postural
Drenaxe postural Dedos achafarrados
Dedos achafarrados Fibrose quística
Fibrose quística
Donaldson SH, Pilewski JM, Griese M, et al. Tezacaftor / ivacaftor en suxeitos con fibrosis quística e F508del / F508del-CFTR ou F508del / G551D-CFTR. Am J Respir Crit Care Med. 2018; 197 (2): 214-224. PMID: 28930490 pubmed.ncbi.nlm.nih.gov/28930490/.
Eagan ME, Schechter MS, Voynow JA. Fibrose quística. En: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Manual de Pediatría. 21a ed. Filadelfia, PA: Elsevier; 2020: cap 432.
Farrell PM, White TB, Ren CL, et al. Diagnóstico da fibrosis quística: pautas consensuadas da Fundación para a fibrose quística. J Pediatr. 2017; 181S: S4-S15.e1. PMID: 28129811 pubmed.ncbi.nlm.nih.gov/28129811/.
Graeber SY, Dopfer C, Naehrlich L, et al. Efectos da terapia con lumacaftor / ivacaftor sobre a función CFTR en pacientes homocigóticos Phe508del con fibrosis quística. Am J Respir Crit Care Med. 2018; 197 (11): 1433-1442. PMID: 29327948 pubmed.ncbi.nlm.nih.gov/29327948/.
Grasemann H. Fibrosis quística. En: Goldman L, Schafer AI, eds. Medicina Goldman-Cecil. 26a ed. Filadelfia, PA: Elsevier; 2020: cap 83.
Rowe SM, Hoover W, Solomon GM, Sorscher EJ. Fibrose quística. En: Broaddus VC, Mason RJ, Ernst JD, et al., Eds. Libro de texto de medicina respiratoria de Murray e Nadel. 6a ed. Filadelfia, PA: Elsevier Saunders; 2016: cap 47.
Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor-ivacaftor en pacientes con fibrosis quística homocigota para phe508del. N Engl J Med. 2017; 377 (21): 2013-2023. PMID: 29099344 pubmed.ncbi.nlm.nih.gov/29099344/.
Artigos De Portal

Como usar a tempada Enerxía de Tauro para adestrar de xeito máis intelixente