Como recoñecer o síndrome de Rett

Contido
- Características do síndrome de Rett
- Como se fai o diagnóstico
- Esperanza de vida
- Que causa o síndrome de Rett
- Tratamento da síndrome de Rett
A síndrome de Rett, tamén chamada hiperamonemiaemia cerebro-atrófica, é unha enfermidade xenética rara que afecta ao sistema nervioso e afecta case exclusivamente ás nenas.
Os nenos coa síndrome de Rett deixan de xogar, quedan illados e perden as habilidades aprendidas, como camiñar, falar ou incluso mover as mans, orixinando movementos involuntarios das mans característicos da enfermidade.
A síndrome de Rett non ten cura, pero pódese controlar co uso de medicamentos que reducen as crises epilépticas, a espasticidade e a respiración, por exemplo. Pero a fisioterapia e a estimulación psicomotriz son de gran axuda e deben realizarse, preferentemente, a diario.
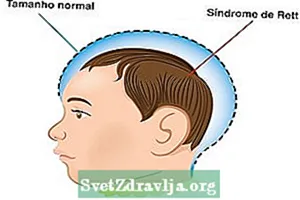
Características do síndrome de Rett
A pesar dos síntomas que máis chaman a atención dos pais só aparecen despois de 6 meses de vida, o bebé con síndrome de Rett ten hipotonía e pode ser visto polos pais e a familia, como un bebé moi "bo" e fácil de coidar. de.
Esta síndrome desenvólvese en 4 fases e ás veces o diagnóstico só chega ao redor dun ano de idade ou máis tarde, dependendo dos signos que presente cada neno.
Primeira fase, ocorre entre os 6 e os 18 meses de vida, e hai:
- Parar o desenvolvemento do neno;
- A circunferencia da cabeza non segue a curva de crecemento normal;
- Diminución do interese por outras persoas ou nenos, con tendencia a illarse.
Segundo nivel, ocorre a partir dos 3 anos e pode durar semanas ou meses:
- O neno chora moito, incluso sen motivo aparente;
- O neno sempre permanece irritado;
- Aparecen movementos repetitivos das mans;
- Aparecen cambios respiratorios, coa respiración detida durante o día, un momento de aumento da frecuencia respiratoria;
- Convulsións convulsivas e ataques de epilepsia ao longo do día;
- Os trastornos do sono poden ser comúns;
- O neno que xa falou pode deixar de falar completamente.


Terceira fase, que ocorre hai aproximadamente 2 e 10 anos:
- Pode haber algunha mellora nos síntomas presentados ata o momento e o neno pode volver a mostrar interese por outros;
- A dificultade para mover o tronco é evidente, hai dificultade para estar de pé;
- A espasticidade pode estar presente;
- Desenvólvese a escoliose que prexudica a función pulmonar;
- É frecuente queixa os dentes durante o sono;
- A alimentación pode ser normal e o peso do neno tamén adoita ser normal, cun lixeiro aumento de peso;
- O neno pode perder a respiración, tragar aire e ter moita saliva.
Cuarta fase, que ocorre arredor dos 10 anos:
- Perda de movemento aos poucos e empeoramento da escoliose;
- A deficiencia mental faise grave;
- Os nenos que foron capaces de camiñar perden esta capacidade e necesitan unha cadeira de rodas.
Os nenos que son capaces de aprender a camiñar aínda teñen algunha dificultade para moverse e, xeralmente, de puntillas ou dan os primeiros pasos cara atrás. Ademais, é posible que non poidan chegar a ningún lado e a súa andaina parece que non ten sentido porque ela non camiña para coñecer a outra persoa, nin para coller xoguetes, por exemplo.
Como se fai o diagnóstico
?????? O diagnóstico faino o neuropediatra que analizará a cada neno en detalle, segundo os signos presentados. Para o diagnóstico débense observar polo menos as seguintes características:
- Aparentemente desenvolvemento normal ata os 5 meses de vida;
- Tamaño normal da cabeza ao nacer, pero non segue a medida ideal despois de 5 meses de vida;
- Perda da capacidade de mover as mans normalmente ao redor dos 24 e 30 meses de idade, dando lugar a movementos incontrolados como torcer ou levar as mans á boca;
- O neno deixa de interactuar con outras persoas ao comezo destes síntomas;
- Falta de coordinación dos movementos do tronco e camiñada descoordinada;
- O neno non fala, non pode expresarse cando quere algo e non entende cando lle falamos;
- Retraso grave no desenvolvemento, con sentado, rastexando, falando e camiñando moito máis tarde do esperado.
Outro xeito máis fiable de descubrir se realmente é esta síndrome é facer unha proba xenética porque aproximadamente o 80% dos nenos coa síndrome de Rett clásica teñen mutacións no xene MECP2. Este exame non o pode facer SUS, pero non o poden negar os plans de saúde privados e, se isto ocorre, ten que presentar unha demanda.
Esperanza de vida
Os nenos diagnosticados con síndrome de Rett poden vivir moito tempo, pasados os 35 anos, pero poden sufrir unha morte súbita durmindo mentres aínda son bebés. Algunhas situacións que favorecen complicacións graves que poden ser mortais inclúen a presenza de infeccións, as enfermidades respiratorias que se desenvolven debido á escoliose e a mala expansión pulmonar.
O neno pode asistir á escola e pode aprender certas cousas, pero o ideal sería integralo na educación especial, onde a súa presenza non chamará a atención, o que podería prexudicar a súa interacción cos demais.
Que causa o síndrome de Rett
O síndrome de Rett é unha enfermidade xenética e normalmente os nenos afectados son os únicos da familia, a non ser que teñan un irmán xemelgo, que é probable que teña a mesma enfermidade. Esta enfermidade non está asociada a ningunha acción que tiveron os pais e, polo tanto, non precisan sentirse culpables.
Tratamento da síndrome de Rett
O pediatra debe levar a cabo o tratamento ata que o neno teña 18 anos e despois debe ser seguido por un médico de cabeceira ou un neurólogo.
As consultas deben realizarse cada 6 meses e neles pódense observar signos vitais, estatura, peso, corrección dos medicamentos, avaliación do desenvolvemento do neno, cambios na pel como a presenza de feridas por decúbito, que son as escaras que poden infectarse. o risco de morte. Outros aspectos que poden ser importantes son a avaliación do desenvolvemento e do sistema respiratorio e circulatorio.
A fisioterapia debe realizarse ao longo da vida da persoa con síndrome de Rett e son útiles para mellorar o ton, a postura, a respiración e pódense usar técnicas como Bobath para axudar ao desenvolvemento do neno.
As sesións de estimulación psicomotriz pódense realizar unhas 3 veces á semana e poden axudar no desenvolvemento motor, diminuír a gravidade da escoliose, o control da baba e a interacción social, por exemplo. O terapeuta poderá indicar algúns exercicios que os pais poden realizar na casa para que o estímulo neurolóxico e motor se realice diariamente.
Ter unha persoa con síndrome de Rett na casa é unha tarefa cansativa e difícil. Os pais poden estar moi drenados emocionalmente e por esta razón poden aconsellarlles que os sigan psicólogos que poidan axudar a tratar as súas emocións.
Asegúrese De Ler

Amiloidose hereditaria
