Síndrome de Angelman
A síndrome de Angelman (SA) é unha enfermidade xenética que causa problemas coa forma en que se desenvolven o corpo e o cerebro dun neno. A síndrome está presente desde o nacemento (conxénita). Non obstante, moitas veces non se diagnostica ata os 6 a 12 meses de idade. É entón cando se notan por primeira vez problemas de desenvolvemento na maioría dos casos.
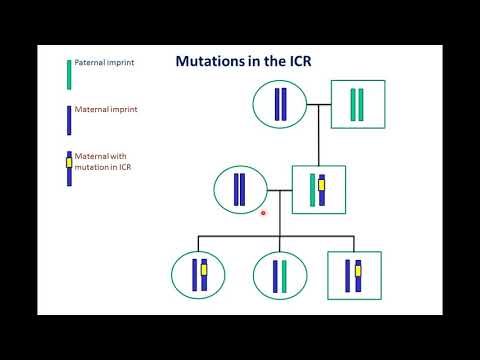
Esta condición implica o xene UBE3A.
A maioría dos xenes veñen en parellas. Os nenos reciben un de cada pai. Na maioría dos casos, ambos os xenes están activos. Isto significa que a información de ambos xenes é utilizada polas células. Co UBE3A xene, ambos os pais transmíteno, pero só o xene transmitido pola nai está activo.
A síndrome de Angelman ocorre con máis frecuencia porque UBE3A transmitido da nai non funciona como debería. Nalgúns casos, AS prodúcese cando hai dúas copias de UBE3A o xene provén do pai e ningún provén da nai. Isto significa que ningún dos dous xenes está activo, porque ambos proceden do pai.
En neonatos e bebés:
- Perda de ton muscular (floppiness)
- Problemas para alimentar
- Azia (refluxo ácido)
- Movemento de brazo e perna tremendo
En nenos pequenos e maiores:
- Camiñando inestable ou desigual
- Pouco ou ningún discurso
- Personalidade feliz e excitábel
- Rir e sorrir a miúdo
- Pelo claro, pel e cor dos ollos en comparación co resto da familia
- Tamaño pequeno da cabeza en comparación co corpo, aplanado na parte traseira da cabeza
- Discapacidade intelectual grave
- Convulsións
- Movemento excesivo das mans e dos membros
- Problemas de sono
- Lingua empurrando, babeando
- Movementos inusuales de mastigación e boca
- Ollos cruzados
- Camiñar cos brazos levantados e as mans ondeando
A maioría dos nenos con este trastorno non presentan síntomas ata uns 6 a 12 meses. É entón cando os pais poden notar un atraso no desenvolvemento do seu fillo, como non gatear ou comezar a falar.
Os nenos de entre 2 e 5 anos comezan a mostrar síntomas como camiñar desigual, personalidade feliz, rir a miúdo, sen fala e problemas intelectuais.
As probas xenéticas poden diagnosticar a síndrome de Angelman. Estas probas buscan:
- Anacos de cromosomas que faltan
- Proba de ADN para ver se as copias do xene de ambos pais están nun estado inactivo ou activo
- Mutación xenética na copia do xene da nai
Outras probas poden incluír:
- Resonancia magnética cerebral
- EEG
Non hai cura para a síndrome de Angelman. O tratamento axuda a xestionar os problemas de saúde e desenvolvemento causados pola enfermidade.
- Os medicamentos anticonvulsivos axudan a controlar as convulsións
- A terapia de conduta axuda a controlar a hiperactividade, os problemas de sono e os problemas de desenvolvemento
- A terapia ocupacional e a fala xestiona problemas de fala e ensina habilidades para vivir
- A fisioterapia axuda a camiñar e a moverse
Fundación Angelman Syndrome: www.angelman.org
AngelmanUK: www.angelmanuk.org
As persoas con SA viven preto dunha vida normal. Moitos teñen amizades e interactúan socialmente. O tratamento axuda a mellorar a función. As persoas con SA non poden vivir sós. Non obstante, poden aprender determinadas tarefas e convivir con outras persoas nun ambiente supervisado.
As complicacións poden incluír:
- Convulsións graves
- Reflujo gastroesofágico (azia)
- Escoliose (columna vertebral curva)
- Lesións accidentais por movementos incontrolados
Chame ao seu médico se o seu fillo ten síntomas desta enfermidade.
Non hai ningunha forma de previr a síndrome de Angelman. Se tes un fillo con SA ou antecedentes familiares da enfermidade, quizais queiras falar co teu provedor antes de quedar embarazada.
Dagli AI, Mueller J, Williams CA. Síndrome de Angelman. GeneReviews. Seattle, WA: Universidade de Washington; 2015: 5. PMID: 20301323 www.ncbi.nlm.nih.gov/pubmed/20301323. Actualizado o 27 de decembro de 2017. Consultado o 1 de agosto de 2019.
Kumar V, Abbas AK, Aster JC. Enfermidades xenéticas e pediátricas. En: Kumar V, Abbas AK, Aster JC, eds. Patoloxía básica de Robbins. 10a ed. Filadelfia, PA: Elsevier; 2018: cap 7.
Madan-Khetarpal S, Arnold G. Trastornos xenéticos e condicións dismórficas. En: Zitelli BJ, McIntire SC, Nowalk AJ, eds. Atlas de diagnóstico físico pediátrico de Zitelli e Davis. 7a ed. Filadelfia, PA: Elsevier; 2018: cap 1.
Nussbaum RL, McInnes RR, Willard HF. A base cromosómica e xenómica da enfermidade: trastornos dos autosomas e cromosomas sexuais. En: Nussbaum RL, McInnes RR, Willard HF, eds. Thompson & Thompson Xenética en Medicina. 8a ed. Filadelfia, PA: Elsevier; 2016: cap 6.
Ler Hoxe

Altitude Enfermidade
