Ventrículo esquerdo de entrada dobre

O ventrículo esquerdo de entrada dobre (DILV) é un defecto cardíaco que está presente desde o nacemento (conxénito). Afecta ás válvulas e ás cámaras do corazón. Os bebés que nacen con esta enfermidade só teñen unha cámara de bombeo en funcionamento (ventrículo) no seu corazón.
O DILV é un dos varios defectos cardíacos coñecidos como defectos do ventrículo único (ou comúns). As persoas con DILV teñen un ventrículo esquerdo grande e un ventrículo dereito pequeno. O ventrículo esquerdo é a cámara de bombeo do corazón que envía ao corpo sangue rico en osíxeno. O ventrículo dereito é a cámara de bombeo que envía sangue con poucos osíxenos aos pulmóns.
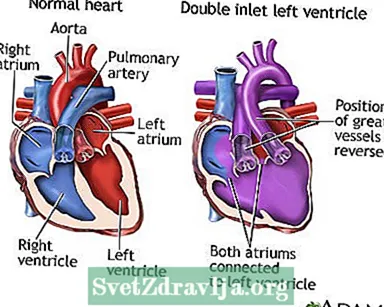
No corazón normal, os ventrículos dereito e esquerdo reciben sangue das aurículas dereita e esquerda. As aurículas son cámaras superiores do corazón.O sangue pobre en osíxeno que volve do corpo flúe cara á aurícula dereita e ao ventrículo dereito. O ventrículo dereito bombea o sangue á arteria pulmonar. Este é o vaso sanguíneo que leva o sangue aos pulmóns para coller osíxeno.
O sangue con osíxeno fresco volve á aurícula esquerda e ao ventrículo esquerdo. A aorta leva entón sangue rico en osíxeno ao resto do corpo desde o ventrículo esquerdo. A aorta é a arteria principal que sae do corazón.
En persoas con DILV, só se desenvolve o ventrículo esquerdo. Ambas aurículas baleiran sangue neste ventrículo. Isto significa que o sangue rico en osíxeno mestúrase con sangue pobre en osíxeno. A mestura é entón bombeada ao corpo e aos pulmóns.
DILV pode ocorrer se os vasos sanguíneos grandes que xorden do corazón están en posicións equivocadas. A aorta xorde do pequeno ventrículo dereito e a arteria pulmonar xorde do ventrículo esquerdo. Tamén pode ocorrer cando as arterias están en posicións normais e xorden dos ventrículos habituais. Neste caso, o sangue flúe do ventrículo esquerdo a dereito a través dun orificio entre as cámaras chamado defecto septal ventricular (VSD).
DILV é moi raro. Descoñécese a causa exacta. O problema ocorre con máis probabilidade no inicio do embarazo, cando se desenvolve o corazón do bebé. As persoas con DILV a miúdo tamén teñen outros problemas cardíacos, como:
- Coarctación da aorta (estreitamento da aorta)
- Atresia pulmonar (a válvula pulmonar do corazón non se forma correctamente)
- Estenose da válvula pulmonar (estreitamento da válvula pulmonar)
Os síntomas de DILV poden incluír:
- Cor azulada na pel e nos beizos (cianose) debido ao baixo osíxeno no sangue
- Non engordar e medrar
- Pel pálida (palidez)
- Mala alimentación por cansarse facilmente
- Suando
- Pernas ou abdome inchados
- Problemas para respirar
Os signos de DILV poden incluír:
- Ritmo cardíaco anormal, como se ve nun electrocardiograma
- Acumulación de fluído ao redor dos pulmóns
- Insuficiencia cardíaca
- Bruxo do corazón
- Ritmo cardíaco rápido
As probas para diagnosticar DILV poden incluír:
- Radiografía de tórax
- Medición da actividade eléctrica no corazón (electrocardiograma ou ECG)
- Exame ecográfico do corazón (ecocardiograma)
- Pasar un tubo fino e flexible ao corazón para examinar as arterias (cateterismo cardíaco)
- Resonancia magnética cardíaca
A cirurxía é necesaria para mellorar a circulación sanguínea a través do corpo e nos pulmóns. As cirurxías máis comúns para tratar DILV son unha serie de dúas a tres operacións. Estas cirurxías son similares ás empregadas para tratar a síndrome do corazón esquerdo hipoplásico e a atresia tricúspide.
A primeira cirurxía pode ser necesaria cando o bebé ten só uns días. Na maioría dos casos, o bebé pode ir a casa dende o hospital despois. O neno normalmente terá que tomar medicamentos todos os días e ser seguido de preto por un médico cardíaco pediátrico (cardiólogo). O médico do neno determinará cando se debe facer a segunda etapa da cirurxía.
A seguinte cirurxía (ou primeira cirurxía, se o bebé non precisou un procedemento como un recentemente nado) chámase procedemento bidireccional de Glenn ou Hemifontan. Esta cirurxía normalmente faise cando o neno ten entre 4 e 6 meses.
Mesmo despois das operacións anteriores, o neno aínda pode parecer azul (cianótico). O último paso chámase procedemento Fontan. Esta cirurxía faise máis a miúdo cando o neno ten entre 18 meses e 3 anos. Despois deste último paso, o bebé xa non é azul.
A operación Fontan non crea unha circulación normal no corpo. Pero mellora o fluxo sanguíneo o suficiente para que o neno poida vivir e medrar.
Un neno pode necesitar máis cirurxías por outros defectos ou prolongar a supervivencia mentres espera o procedemento de Fontan.
É posible que o seu fillo precise tomar medicamentos antes e despois da cirurxía. Estes poden incluír:
- Anticoagulantes para evitar a coagulación do sangue
- Inhibidores de ECA para reducir a presión arterial
- Digoxina para axudar a contraer o corazón
- Pílulas de auga (diuréticos) para reducir o inchazo no corpo
Pódese recomendar un transplante de corazón se fallan os métodos anteriores.
DILV é un defecto cardíaco moi complexo que non é doado de tratar. O ben que o faga o bebé depende de:
- O estado xeral do bebé no momento do diagnóstico e tratamento.
- Se hai outros problemas cardíacos.
- Que grave é o defecto.
Despois do tratamento, moitos bebés con DILV viven para ser adultos. Pero requirirán seguimentos ao longo da vida. Tamén poden sufrir complicacións e poden ter que limitar as súas actividades físicas.
As complicacións de DILV inclúen:
- Paliza (engrosamento das camas das uñas) nos dedos dos pés e dos dedos (sinal tardío)
- Insuficiencia cardíaca
- Pneumonía frecuente
- Problemas no ritmo cardíaco
- Morte
Chame ao seu médico se o seu fillo ou filla:
- Parece cansarse facilmente
- Ten problemas para respirar
- Ten a pel ou os beizos azulados
Fale tamén co seu provedor se o seu bebé non medra nin engorda.
Non hai prevención coñecida.
DILV; Ventrículo único; Ventrículo común; Corazón univentricular; Corazón univentricular do tipo ventricular esquerdo; Defecto cardíaco conxénito - DILV; Defecto cardíaco cianótico - DILV; Defecto de nacemento - DILV
 Ventrículo esquerdo de entrada dobre
Ventrículo esquerdo de entrada dobre
Kanter KR. Manexo de conexións do ventrículo único e cavopulmonar. En: Sellke FW, del Nido PJ, Swanson SJ, eds. Cirurxía do peito de Sabiston e Spencer. 9a ed. Filadelfia, PA: Elsevier; 2016: cap 129.
Kliegman RM, St. Geme JW, Blum NJ. Shah SS, Tasker RC, Wilson KM. Schor NF. Cardiopatía conxénita cianótica: lesións asociadas ao aumento do fluxo sanguíneo pulmonar. En: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Manual de Pediatría. 21a ed. Filadelfia, PA: Elsevier; 2020: cap 458.
Wohlmuth C, Gardiner HM. O corazón. En: Pandya PP, Oepkes D, Sebire NJ, Wapner RJ, eds. Medicina fetal: ciencia básica e práctica clínica. 3a ed. Filadelfia, PA: Elsevier; 2020: cap 29.
Publicacións Interesantes

Coidados dentais - adultos
